My name is Gwydyon Marchelli. I was born the 11/11/1992 in Turin (Italy) and I lived in a small town near my birthplace named San Benigno Canavese. I studied at the University of Turin where in the 2015 I get my bachelor degree In Chemistry and Chemical Technologies. I did an internship in the University of Pau from May 2017 to July 2017 and in April 2018 I get my master degree in Chemistry at the University of Turin.
My name is Gwydyon Marchelli. I was born the 11/11/1992 in Turin (Italy) and I lived in a small town near my birthplace named San Benigno Canavese. I studied at the University of Turin where in the 2015 I get my bachelor degree In Chemistry and Chemical Technologies. I did an internship in the University of Pau from May 2017 to July 2017 and in April 2018 I get my master degree in Chemistry at the University of Turin.
During my studies I discover an interest for the theoretical chemistry, in particular the use of the computational tools in order to understand the behavior of the solid state. I focused on the optical properties of it; nevertheless my preparation includes also organic, inorganic systems and instrumental analysis.
Now I am the ESR 10 in the etn SOCRATES, doing the PhD at the University of Bonn with the project “In-silico design of selective metal extractants”. If you are interested in my works don’t hesitate to contact me at marchelli@thch.uni-bonn.de
Introduction and objectives
Understanding the ion-solvent interaction is a fundamental step in order to perform the extraction of the so-called critical metals from liquid phase. Those kinds of interactions are often complicated to visualize and to investigate via experimental set-ups. For this reason a computational approach is required.
In the PhD, I focused on the simulation of the liquid phase with different methods, which will be briefly described in the next section. In particular I investigated the properties of binary mixtures, the hydrogen bond network in small alcohols and the coordination of transition metal in deep eutectic solvents (DES).
The objectives are the following:
- To investigate the metal-ionic liquids interactions, and defining possible ways to introduce selectivity;
- To identify lead compounds for the selective extraction of 4 different metals.
Methods
There are two main approaches to model the liquid phase:
- Quantum mechanical methods, which solve the Schroedinger equation to find all the possible information about the investigated systems. Since the exact solution cannot be achieved other than for hydrogenoid atoms, approximations of the Hamiltonian are necessary. Different approaches and different levels of theory are available. I mainly used the Density Functional Theory (DFT) method, along with dispersion corrections and the triple Z wave function basis set. Usually this level of theory is called ab-initio or first principle.
- As an alternative, a pre-defined potential can be used, where empirically fitted data are taken into account. Different force fields were calculated in the last decades to describe different properties and structures.
Quantum mechanical calculations are carried out as statical gas-phase like simulation via programs such as ORCA and Turbomole.
The liquid phase is simulated through molecular dynamics simulation (both classical, so at the force field level of theory, and ab-initio) as well as via quantum cluster equilibrium (QCE) approach, where it is described as an ensemble of smaller clusters.
Results obtained
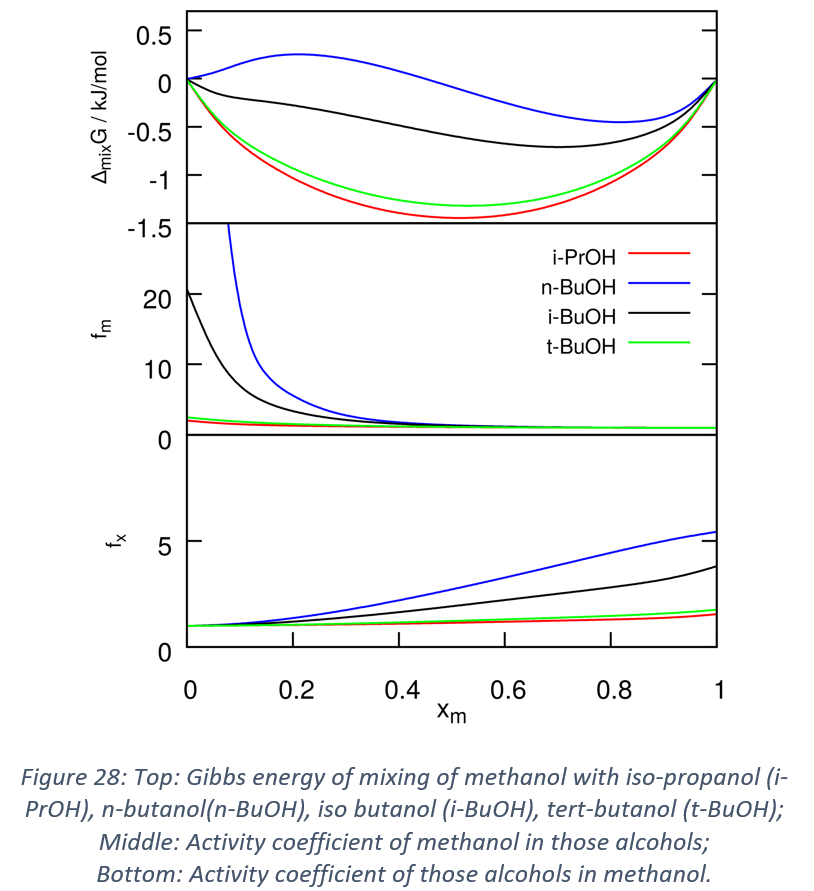
- Activity coefficients have been calculated via QCE for different binary mixtures. They are good indicators of the deviation with respect to the ideal situation for mixtures. First this approach has been evaluated for well-known and representative systems. More recently, I have used the activity coefficients analysis to assess the effects in small alcohols of branching and of an increasing side chain size for small alcohols. The results are promising, and it is clear that when increasing the branching, the system deviates less from the ideal situation, while increasing the side chain leads to the opposite effect. These results are presented in Figure 27 and they are summarized in a submitted article.
- The coordination of iron in deep eutectic solvents (choline chloride with ethylene glycol, ratio 1:2) has been investigated via ab-initio molecular dynamics. All the possible spin states are considered for both Fe2+ and Fe3+. From Figure 28 it is clear how the coordination can drastically change with respect to the spin state. These are still preliminary results and further investigations are required to confirm these considerations and proceed further in the analysis of the systems.

Conclusion & Outlook for further work
In the last 19 months, I have focused on the investigation of the hydrogen bond network of different mixtures, in order to evaluate novel strategies to extract critical metals from a liquid phase. I have proved that the hydrogen bond network is influenced by the size and branching of the hydrogen bond donor and acceptor, and this result will influence the modelling of novel hydrophobic deep eutectic solvents. At the same time, I have also investigated the coordination of metals in well-known deep eutectic solvents.